
 The FDA’s Title 21 CFR 820 Regulatory Requirement means that manufacturers of medical devices that are subject to FDA regulation need to establish and maintain procedures for implementing corrective and preventive action (CAPA) related to device non-conformity.
The FDA’s Title 21 CFR 820 Regulatory Requirement means that manufacturers of medical devices that are subject to FDA regulation need to establish and maintain procedures for implementing corrective and preventive action (CAPA) related to device non-conformity.
These procedures must provide for control and action to be taken on any distributed or not yet distributed devices that are suspected of having potential nonconformities.
According to the FDA, plans with respect to implementing CAPA should include:
- Establishing data sources and criteria
- Measuring and analysis of data sources
- Improvement plans
- Input to management
Let’s look at several areas of FDA guidance. For each, I will point out where the right CRM system, properly configured, can facilitate data collection, analysis and reporting.
CAPA Related Data Collection
CAPA conformity has an important data collection element. To the extent that data can be gathered into a single repository (rather than into separate spreadsheets), data analysis and reporting are less time consuming and more efficient. Results are more easily shareable.
External Data Sources
In its guidance for CAPA, the FDA has identified the following set of external data sources.
- Supplier controls
- Customers
- Complaints
- Servicing repairs
- Adverse event reporting (MDR)
- FDA
- Similar devices from competitors
Managing Complaints, Service Issues and Repairs With CRM
 CRM is an efficient platform for collecting and aggregating multiple types of external data inputs. Several external data sources cited by the FDA can be managed under the umbrella of CRM case management functionality.
CRM is an efficient platform for collecting and aggregating multiple types of external data inputs. Several external data sources cited by the FDA can be managed under the umbrella of CRM case management functionality.
Once a CRM system is configured for case management and the users are trained, external data can be automatically or manually entered into the system.
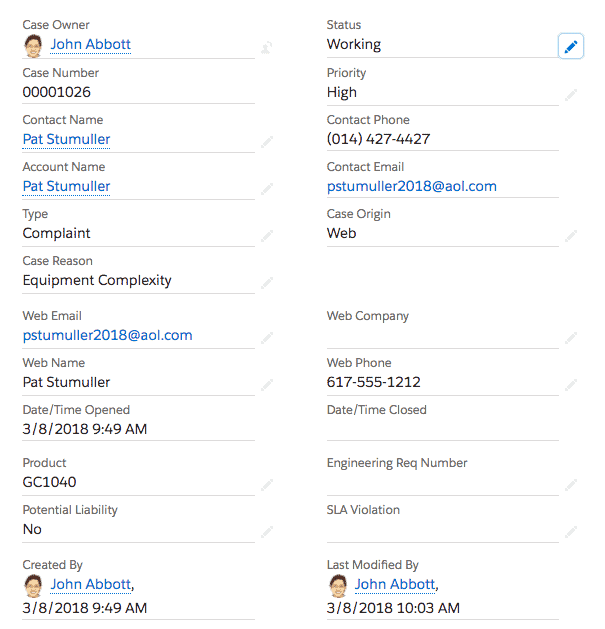
The FDA defines “complaint” as any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution.
With CRM, a number of electronic submission options are available.
An email to an address such as complaint@manufacturer.com can immediately create a new complaint record in a CRM system. A complaint can be submitted directly into a CRM system via a web form.
When submitted service issues and complaints are properly categorized within a CRM system, reporting and dashboards expose the most common types of complaints and issues.
Each time a complaint or issue is logged in a CRM system, the complaint or service issue can be classified by a CRM user. A common user interface means that anyone who logs or manages any complaint or issue within a CRM is presented with the same set of classification choices.
When a case is created in CRM for a service repair, the type of problem and the corrective action or repair can be logged by a technician.
CRM and Complaint Handling Procedures
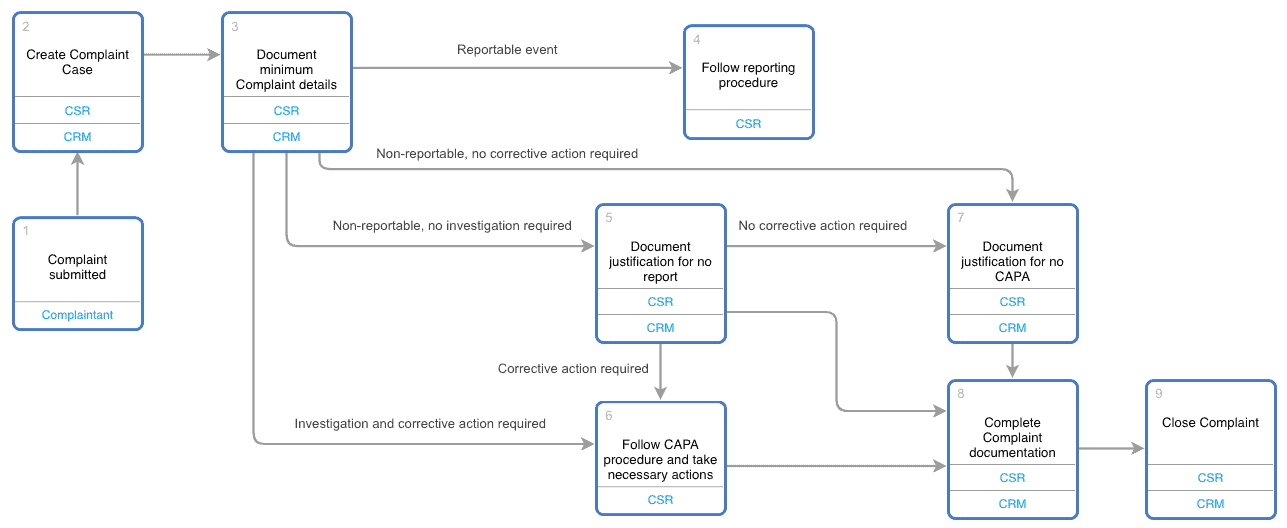
A CRM system can be configured to support a complaint handling procedure. The CRM system workflows can be reflective of appropriate procedure flow diagrams. Flow diagrams themselves can be a part of the CRM user interface for easy user reference.
As a specific CRM vendor example, Elements.cloud flow diagrams can be embedded in Salesforce Complaint and Service records.
Data Analysis
 The FDA requires that procedure include requirements for, among other things, “service records, complaints, returned product, and other sources of quality data…”
The FDA requires that procedure include requirements for, among other things, “service records, complaints, returned product, and other sources of quality data…”
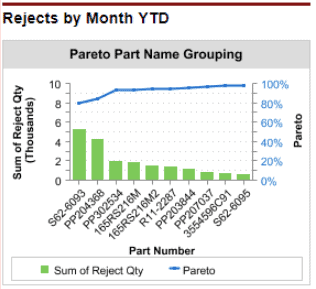
According to the FDA, “the analysis of product and quality problems should include appropriate statistical and non-statistical techniques. Statistical techniques include Pareto analysis, spreadsheets, and pie charts.”
Spreadsheets work well for data analysis, but do not have strong data collection mechanisms. A CRM system works well for both data collection and analysis.
Pareto charts in a CRM system can display the most common types of service issues, complaints and reasons for product returns. Salesforce is a CRM system that allows for creation of Pareto charts as part of a dashboard.
Corrective Action Documentation
CAPA actions taken must be thoroughly documented for regulatory purposes. In fact, all activities under the FDA’s above mentioned Title 21 CFR 820 Regulatory Requirement and their results are required to be documented.
While medical device manufacturers have a number of choices as to where to centrally store documentation, a CRM system document management area is certainly an option.
Input to Management
The FDA emphasizes that it is always management’s responsibility to ensure that all nonconformity issues are handled appropriately.
Relevant information on identified quality problems, as well as corrective and preventive actions should be submitted for management review.
Reports for certain facets of reported quality problems, such as information that is cataloged within CRM cases, can be outputted via CRM reports or document merges.
An FDA Inspection
The FDA states, “Manufacturers should consider that their Corrective Action and Preventive Action documentation can demonstrate to FDA that the manufacturer’s quality system is effective and enables the manufacturer to identify problems quickly and implement effective corrective and preventive actions.”
A properly configured CRM system can be a component of what is demonstrated to an FDA inspector, should an inspection take place.
Free Guide for Health and Life Science Organizations
Learn 12 ways medical device companies can maximize return on their CRM investment

